论文Learning the rules of peptide self-assembly through data mining with large language models(《通过大型语言模型的数据挖掘学习肽自组装的规则》)系统性研究了肽的自组装行为,重点在于结合实验数据和机器学习技术构建肽自组装行为的预测模型,并开发了一种基于大语言模型(LLM)的高效文献挖掘工作流。
论文作者为Zhenze Yang, Sarah K. Yorke, Tuomas P. J. Knowles, Markus J. Buehler,来自MIT和University of Cambridge。
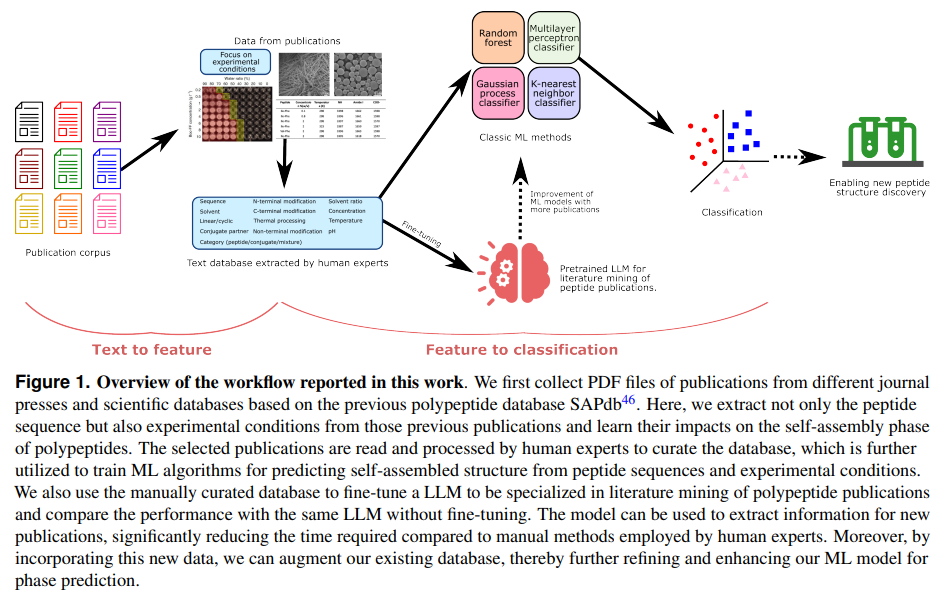
一、背景与研究动机
肽自组装(peptide self-assembly)是指肽分子通过弱的非共价相互作用(如氢键、范德华力等)自行聚集、排列成特定的结构。肽自组装在生物材料、纳米技术、药物递送、组织工程等领域具有重要应用。然而,肽自组装的行为非常复杂,受多种内在和外在因素(如肽序列、环境条件、溶剂类型、pH值、温度等)的影响。尽管已有大量实验研究探讨了这些因素对肽自组装的影响,但目前缺乏一个系统化的研究,能够将这些分散的实验数据整合起来,并全面总结影响肽自组装行为的基本规则。因此,研究团队希望通过结合机器学习(ML)与大语言模型(LLM)来解决这一问题。
二、主要贡献
- 数据收集:构建了一个包含超过1,000条数据的肽自组装数据库,其中包含肽序列、实验条件、组装相态等信息。
- 机器学习模型:基于上述数据,训练了多种机器学习模型(如随机森林、支持向量机等),并应用于预测肽的自组装相态。
- 大语言模型:开发了一个基于大语言模型(如GPT-3.5)的文献挖掘系统,从文献中高效提取实验数据,为肽自组装的研究提供新的工具。
- 研究工具的创新:提出了一个结合人工与LLM的高效数据挖掘流程,能够大大提高文献数据提取的效率,减少人工工作量。
三、方法与实验设计
1. 数据收集与数据库建设
- 数据源:从已存在的SAPdb数据库(包括1,049条记录)中筛选出符合实验条件的文献数据。SAPdb主要包含了短肽(如二肽、三肽)的自组装数据。
- 数据筛选:筛选出75篇文献,提取其中包含肽序列、溶剂、温度、pH、浓度等实验参数的数据。最终构建了一个包含1,012条数据的数据库。
2. 数据处理与特征设计
- 分类特征:包括肽序列、N端修饰、C端修饰、非末端修饰、是否是线性肽或环状肽、肽系统的类型(肽/共轭物/混合物)等9个分类特征。这些特征通常是通过人工标注和文本挖掘得到的。
- 数值特征:包括溶剂比率、肽浓度、pH值、温度等4个数值特征。所有数值数据都进行了归一化处理,以便输入到机器学习模型中进行训练。
- 数据分布:数据的自组装相态分布如下:
- 最常见的相态:包括“无组装”(no-assembly),这一类数据是训练机器学习模型时非常关键的“负样本”。
- 其他自组装相态:如水凝胶(hydrogel)、纤维(fiber)、球形(sphere)等。
3. 机器学习模型训练与优化
- 模型选择:使用了四种经典的机器学习模型:
- 随机森林(Random Forest,RF)
- 多层感知器(Multilayer Perceptron,MLP)
- 高斯过程分类器(Gaussian Process Classifier,GPC)
- K最近邻(K-Nearest Neighbor,KNC)
- 性能评估指标:使用精度(precision)、召回率(recall)和F1分数来评估模型的表现。在所有模型中,随机森林(RF)模型的表现最为优越,达到了精度0.814、召回率0.806、F1分数0.808。
- 数据不平衡处理:由于数据集中某些相态(如“无组装”)的数据量较大,而其他相态的数据量较少,采用了SMOTE(Synthetic Minority Oversampling Technique)算法对数据进行了过采样,以平衡各个类别的样本数量。
- 特征重要性分析:使用SHAP(SHapley Additive exPlanations)方法分析了输入特征对模型预测结果的影响。发现“溶液环境”(solution)、N端修饰(N-terminal modification)和肽浓度(concentration)是最重要的影响因素。
4. 大语言模型(LLM)辅助的文献挖掘
- 文献数据挖掘流程:
- 文献获取:从75篇相关文献中提取文本数据,使用PDFMiner工具将PDF文件转换为文本。
- 实验部分筛选:根据文章的标题和段落标头(如“Material(s)”、“Method(s)”、“Experimental section”)筛选出包含实验细节的段落。
- 关键词筛选:通过设定关键词(如“hydrogel”、“fiber”等)筛选出包含自组装信息的段落。
- 模型微调:将手动构建的数据库(包含52篇文献的训练集与23篇文献的测试集)用作微调GPT-3.5模型的基础。通过微调,模型能够显著提高从文献中提取信息的准确性,尤其在分类特征的提取上,微调后的模型的准确率达到了80%以上,而原始模型只有62.7%。
四、结果
- 机器学习模型性能:通过比较不同算法的表现,RF模型表现最佳,且在数据不平衡和泛化能力上也表现稳定。
- 文献挖掘效率:微调后的GPT模型显著提高了文献数据提取的效率,尤其在提取复杂的实验条件和自组装相态信息时,能够大大减少人工处理的工作量。
- 模型解释性:通过SHAP分析,研究揭示了不同实验条件对肽自组装行为的影响,尤其是肽浓度和溶液环境对自组装相态的关键作用。
五、讨论
- 自组装相态的预测:研究表明,机器学习模型可以有效地根据肽序列和实验条件预测肽的自组装相态。通过模型的特征重要性分析,可以指导实验设计,选择合适的实验条件,以实现期望的自组装结构。
- 大语言模型的应用:LLM的应用大大提高了文献数据的提取效率,尤其是在处理长篇文献时,微调后的LLM能更好地提取实验细节,进而加速数据集的构建。
- 局限性与挑战:尽管该研究提出了一个有效的工作流,但仍存在一些挑战:
- 文献中某些数据缺失或格式问题(如没有详细实验条件或表格信息)会影响数据的完整性;
- 数据集中的一些肽序列或实验方法过于集中于少数几类,这可能导致数据的偏倚。
六、未来展望
- 改进数据多样性:未来可以通过高通量筛选或主动学习等方法增加数据集的多样性,尤其是增加未被充分研究的肽序列和实验条件。
- 生成模型应用:研究可以考虑使用生成模型(如变分自编码器、生成对抗网络等)来生成新的实验数据,进一步丰富数据库。
- 跨学科整合:未来的研究可以结合更多领域的知识(如计算化学、材料科学等),使得肽自组装的预测模型能够更准确地考虑不同领域的实验条件和机制。
Peptideminer on GitHub: https://github.com/lamm-mit/peptideminer