单细胞RNA测序(scRNA-seq)革新了我们对细胞多样性的理解,然而,现有的单细胞基础模型(scFMs)在可扩展性、对多样任务的灵活性以及原生整合文本信息的能力方面仍然存在局限。论文Scaling Large Language Models for Next-Generation Single-Cell Analysis基于 Cell2Sentence(C2S)框架——该框架将scRNA-seq表达谱表示为文本形式的“细胞句子(cell sentences)”——构建了用于单细胞分析的大型语言模型(LLMs),训练语料包含超过十亿个token,由转录组数据、生物文本和元数据组成。通过将模型规模扩展至270亿参数,观察到其在预测与生成能力、以及处理需要跨多细胞语境合成信息的高级下游任务方面均有持续改进。借助现代强化学习技术进行的针对性微调,论文提出的方法在扰动响应预测、自然语言解释和复杂生物推理等任务中表现优异。通过前所未有的规模整合转录组和文本数据,该方法不仅超越了专门的单细胞模型与通用语言模型,还奠定了下一代单细胞分析的强大平台基础,为构建“虚拟细胞(virtual cells)”系统铺平了道路。
论文作者为Syed Asad Rizvi, Daniel Levine, Aakash Patel, Shiyang Zhang, Eric Wang, Sizhuang He, David Zhang, Cerise Tang, Zhuoyang Lyu, Rayyan Darji, Marlene Li, Emily Sun, David Jeong, Lawrence Zhao, Jennifer Kwan, David Braun, Brian Hafler, Jeffrey Ishizuka, Rahul Dhodapkar, Hattie Chung, Shekoofeh Azizi, Bryan Perozzi和David van Dijk,来自Yale University和Google。
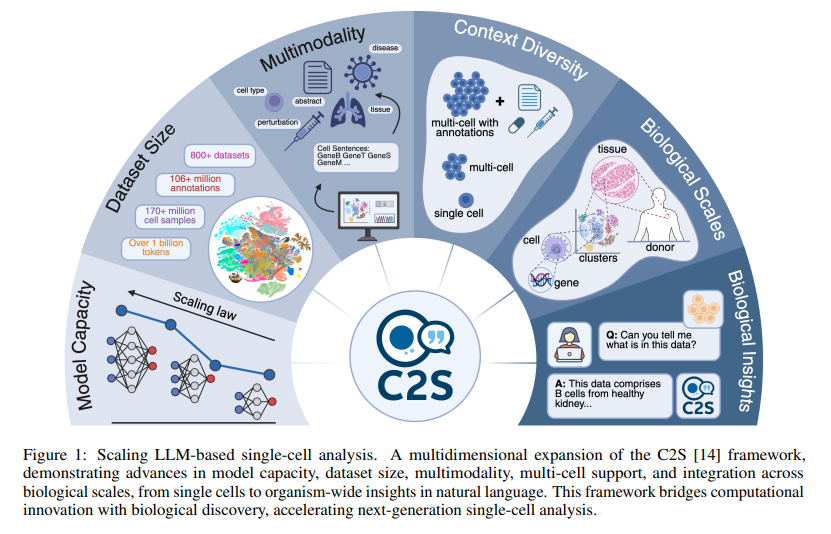
一、研究背景与科学挑战
单细胞RNA测序(single-cell RNA sequencing, scRNA-seq)技术使我们能够以单个细胞为单位,对基因表达谱进行测量,揭示细胞异质性、生物过程状态转变、细胞命运决定等生物学现象。近年来,scRNA-seq数据爆炸式增长,构建了如Human Cell Atlas、CellxGene等全球规模的细胞图谱。
然而,如何利用这些海量scRNA-seq数据进行高效、通用的建模与推理仍面临诸多挑战:
- 传统scRNA-seq建模依赖于“表达矩阵+下游工具”的范式,工具间缺乏统一接口;
- 现有单细胞基础模型(如scGPT、Geneformer、scFoundation)大多采用定制模型结构,缺乏对多模态信息(如文本、生物注释、实验条件等)的融合能力;
- 多数模型无法进行自然语言交互、泛化能力不足;
- 任务范围狭窄,多局限于聚类、降维、注释等传统流程,难以支持生成、问答、复杂条件推理。
在自然语言处理领域,大型语言模型(LLMs)已展现出在统一建模、多任务泛化、语义理解等方面的非凡能力。若能将scRNA-seq问题表征为“文本问题”,即可利用LLMs的强大能力实现scRNA-seq分析范式的革命性转变。
二、方法框架:C2S-Scale 的设计理念
本研究提出 C2S-Scale 框架,是在Cell2Sentence(C2S)思想基础上的扩展,核心是将高维基因表达谱通过降序排序转化为自然语言形式的“细胞句子”(cell sentence),再利用通用LLM进行训练、推理和生成,整体设计思路包括:
- 数据转换:每个细胞的表达向量 X∈RD 中,选取表达量前K高的基因,按表达量降序排列其基因名,构成形如 “CD4 PTPRC IL7R…” 的文本序列。
- 多模态数据融合:将scRNA-seq表达数据、元数据(如组织来源、疾病状态、细胞类型)、自然语言文本(如论文摘要)合并为多模态训练语料。
- 使用多种规模LLM:使用从410M到270亿参数(410M, 1B, 2B, 9B, 27B)的模型,基于Gemma-2和Pythia架构。
- 多任务预训练+任务微调:包括细胞类型注释、条件生成、数据集摘要、空间位置推理、问题解答等任务。
- 使用强化学习(GRPO)进一步对齐输出与生物学偏好,提升自然语言问答与生成任务的准确性和解释力。
三、训练数据构建与语料设计
构建了一个超大规模的多模态训练语料:
- 数据来源:50,000,000+ 人类与小鼠细胞表达数据,来自CellxGene、Human Cell Atlas等开放数据库;
- 数据内容:每个细胞包含表达谱、注释(细胞类型、组织、疾病状态、供体ID、物种、发育阶段等)、元数据(文献摘要、标题);
- 转换为文本:将每个细胞转换为cell sentence,同时生成自然语言任务指令;
- 构建任务样本:共构造约1.5亿个样本,涵盖12类任务,包括单细胞建模、多细胞推理、文本生成、元数据预测、条件生成等。
所有任务样本构建时均以自然语言格式组织,例子如下:
- 输入:Cell sentence + Prompt,如“预测该细胞的类型:”;
- 输出:自然语言标签,如“CD8+ T cell”;
- 条件生成输入:组织=“lung”,任务=“生成5个对应细胞”;
- 输出:5个细胞句子文本。
四、模型架构与技术实现细节
- 模型结构:基于Transformer解码器架构,采用Decoder-only结构,符合主流生成式LLM设计(如LLaMA、GPT系列);
- 词嵌入(word embedding):无需引入新词表,直接使用原始tokenizer对基因名进行编码;
- 自注意力机制(Self-attention):用于建模cell sentence内部基因之间的依赖关系,以及细胞间和文本间上下文关系;
- 多任务训练目标:
- 基础预训练目标为下一个token预测(Next Token Prediction);
- 微调阶段也采用相同目标,确保任务统一性;
- 参数高效训练:
- 小模型(≤1B)使用PyTorch + Huggingface;
- 大模型(2B–27B)在TPU+JAX上训练;
- 微调阶段引入LoRA(Low-Rank Adaptation)与adapter技术;
- 强化学习方法:
- 使用Group Relative Policy Optimization(GRPO);
- 奖励函数为BioBERTScore,衡量生成结果与参考答案在生物语义空间的一致性;
- 策略更新基于候选输出的相对得分,而非独立回报函数。
五、核心实验与评估结果
- 模型能力评估:
- 预测任务:细胞类型预测、组织推断;
- 生成任务:条件生成、扰动响应生成;
- 多模态理解:自然语言摘要、问答;
- 空间推理:预测空间邻域结构、基于CellPhoneDB和BioGRID的基因交互;
- 结果展示:
- C2S-Scale在所有任务上均显著优于scGPT、scGen、CellOT、Geneformer等;
- 与通用LLM如GPT-4o、Gemini、LLaMA系列相比,在领域任务上也占据优势;
- 支持多细胞输入,实现更强的空间上下文建模与条件生成;
- 模型扩展性:
- 随着参数数增加(410M→27B),性能线性提升;
- 训练样本数增加也带来稳定收益;
- 小模型(如2B)通过LoRA也可实现优良表现;
- 新指标:scFID
- 基于scFM嵌入空间的Frechet Distance;
- 替代传统MSE或expression-level指标,鲁棒性更强,更贴近生物意义;
- 特别适用于生成任务的定量评估。
六、任务实例与具体结果详述
- Cluster Captioning:用GPT-4生成训练数据,C2S-Scale生成细胞群描述;
- 测试指标:BioBERTScore;
- 在未见过的数据集上也能准确描述主细胞类型、组织来源、疾病状态;
- Dataset Interpretation:
- 输入:随机采样多个细胞句子;
- 输出:类似生物摘要的段落;
- 模型可泛化至完全未见数据集,准确总结主要信息;
- Perturbation Prediction:
- Dong等数据集:细胞因子组合刺激实验;
- L1000数据集:药物诱导的凋亡表达模式;
- 采用scFID、Wasserstein、MMD等指标,C2S-Scale全面优胜;
- RL进一步显著提升干扰反应建模的准确性与泛化性;
- Question Answering:
- 构造2000个scQA样本:包括背景信息、数据、问题、答案;
- 比较模型:C2S-Scale > GPT-4o > LLaMA > BioMistral;
- 使用GRPO训练提升答案质量与一致性;
- 能回答如“哪些基因对IFN-β刺激最敏感?”等复杂问题。
七、方法优势与科学创新
- 第一次实现自然语言与scRNA-seq数据的原生统一建模;
- 跨模态、跨尺度建模:从单细胞、细胞群到数据集、器官乃至实验全景;
- 利用通用LLM的强大迁移能力,无需定制神经网络结构;
- 基于数据工程的转换(cell sentence),可拓展至蛋白质组、甲基化组等领域;
- 融合RL对输出进行语义层级的对齐,保障生成结果质量;
- 具有开放性:论文承诺开放模型权重、预训练语料、任务格式,促进社区采纳与研究。
八、局限性与未来方向
- Causal Attention限制:
- 因果注意力机制可能妨碍从低表达基因向高表达建模;
- 可通过引入Bidirectional注意力、随机打乱排序训练等策略改善;
- 模型“幻觉”问题:
- 尽管用于结构化任务表现优越,但在抽象生成、问答中仍可能输出“生物上合理但错误”的结果;
- 后续需引入生物验证器或置信估计;
- 尚未整合蛋白质组、临床数据等更复杂模态;
- 尚未解决解释性问题(为何模型认为该细胞为T细胞?);
- 模型训练资源消耗高,需发展轻量模型与推理加速方案。
九、结论与未来展望
C2S-Scale 框架代表了单细胞分析方法论的重大飞跃,其通过将高维生物数据转化为自然语言,使得通用大型语言模型可直接用于细胞表达建模与生物推理。其优势体现在:
- 通用性强:可迁移至不同生物领域与任务;
- 模型能力强大:覆盖预测、生成、推理全谱任务;
- 接口友好:支持自然语言提示,便于用户交互;
- 模型开放:促进研究复现与再开发。
未来,随着数据模态的不断丰富(如空间组学、时间序列组学、图谱组学等)、LLM结构与对齐方法的不断优化,C2S-Scale 有望成为支持个性化医学、药物开发、疾病建模、临床辅助决策的核心引擎之一,为构建“虚拟细胞系统”与“语言驱动的生物发现平台”打下坚实基础。